|
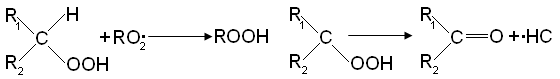
Реферат: Окисление парафиновых углеводородовРеферат: Окисление парафиновых углеводородовОкисление парафиновых углеводородов Ряд процессов жидкофазного окисления углеводородов в настоящее время реализованы как крупнотоннажный производства, например СЖК [2], высших жирных спиртов и др. [3]. Изучения жидкофазного окисление насыщенных углеводородов оказалось весьма плодотворным для установления общих закономерностей процесса окисления. На примере окисления индивидуальных углеводородов и их смесей получены фундаментальные знания о механизме радикальных реакций [3,4-7]. Некоторые особенности процесса жидкофазного окисления парафиновых углеводородов. Окисления парафиновых углеводородов хорошо изученный процесс [7,8,10-12]. Известно, что окисление парафиновых углеводородов молекулярным кислородом приводит к образованию большого число промежуточных и конечных кислородсодержащих продуктов: перекисей, спиртов, карбонильных соединений, кислот, эфиров, а также бифункциональных соединений. Найден ряд катализаторов процесса окисления углеводородов, таких как, растворимые комплексы титана [9], хлорид платины [14], комплексы ванадия (5V) [15], Pd, Pt, Co, Fe нанесенные на носитель, например, на цеолита [16], система на основе Ti, Zr, V, Cr, Mo, W, Mn, Fe и имида [19], система из растворимых соединений кобальта и хрома [21], мультиоксиды металлов [23], алкилперокси- комплексы трехвалентного кобальта [25], смесь азотной кислоты и уксусного ангидрида [26], комплексы марганца и органических кислот содержащих ароматических фрагментов [29], комплексы металлов [30], SiO2 , AI2O3 , ZrO и другие на носителе [31] комплексы металлов, содержащую имидную группировку [32], система на основе Bi, V, Mo, Ag [33], Мn содержащий катализатор, нанесенный на молекулярный сита [34]. Известны каталитические системы ведущие процесс окислению углеводородов селективно [13,18,22,24,28]. К настоящему времени считается доказанным, что в случае окисления предельных углеводородов гидроперекиси единственные первичные промежуточные продукты. Изучения строения образующихся при окислении гидроперекисей показало, что строение углеводородного радикала R в гидроперекиси R'OOH сохраняется таким же, как и в исходном углеводороде RH [3]. Образующиеся при окислении радикалы R'02 взаимодействует с молекулой исходного углеводорода, отрывая атом водорода и образуя гидроперекиси по реакции
При окислении разветвленных парафинов с двумя третичными связями С - Н в большом количестве были обнаружены дигидроперекиси. Окисление проводили при 115 — 120°С до глубины 5 -8 % (мол.) [3]. В начальный период окисления свободные радикалы образуются при взаимодействии исходного углеводорода с растворенным в нем кислородом
Радикал R* присоединят к себе молекулу кислорода и превращается в перекисный радикал RO2•, который далее отрывает атом водорода от молекулы углеводорода и образует гидроперекись и свободный радикал R•, продолжающий цепь. В процессе окисления накапливается гидроперекись, молекулы который сравнительно медленно распадаются на радикалы, например по реакции
Это приводит к увеличению скорости образования свободных радикалов. Процесс распада промежуточных гидроперекисей на радикалы представляет собой реакцию вырожденного разветвления цепей [3]. В целом механизм цепного окисления углеводородов может быть представлен следующим образом [41]:
Имеющийся в настоящее время экспериментальный материал подтверждает цепную схему окисления углеводородов. Чем выше скорость образования свободных радикалов, тем выше их концентрация, тем чаще происходит встреча и рекомбинация (или диспропорционирование) двух свободных радикалов и тем короче цепь обрыв цепей может происходит при взаимодействии свободного радикала со стенкой реактора (обрыв на стенке), а также по бимолекулярной реакции между двумя свободными радикалами (квадратичный обрыв). В жидкой фазе диффузия свободных радикалов у стенке весьма затруднена из - за высокой вязкости среды. По этому в цепных жидкофазных реакциях осуществляется квадратичный обрыв цепей по реакциям:
где: МП - молекулярные продукты. Эти реакции протекают с малой энергией активации, в 4,1 - 8,4 кДж/моль. Реакции между двумя вторичными перекисными радикалами приводит к образованию спирта и кетона. Происходящее в процессе окисления превращение молекулы углеводорода последовательно в гидроперекись, спирт и кетон сохраняет исходный углеводородный скелет молекулы. В процессе окисление кислоты декарбоксилируются сравнительно медленно, и их состав практически не меняется в ходе окисления. Среди кислот, образующихся при окислении н- декана обнаружены окси- и кетокислоты (15- 18% от общего число кислот). Однако эти кислоты образуются не из жирных кислот, а параллельно с ними. Скорость образования уксусной кислоты составляет только 30% от скорости окисления кетона. Следовательно, механизм разрыва а - С - С- связи в окисляющихся парафинах не единственный, и по этому направлению образуются меньше половины низших кислот при окислении парафинов. При большой скорости растворения кислорода его концентрация в окисляющемся веществе близка к насыщению; процесс протекает в кинетической области, т.е. не зависит от скорости растворения и диффузии кислорода в жидкой фазе. При очень быстром окислении диффузия кислорода в жидкость может оказаться лимитирующий стадией процесса окисления. В этом случае реакция будет протекать в диффузионной области. Поэтому, при изучении закономерности реакции окисления протекает в кинетической области [3]. Так как для подавляющего большинство органических соединений, окисляющихся в жидкой фазе, энергия разрыва связи С - Н меньше 377 кДж/моль, то в жидкой фазе зарождение цепей должно происходить преимущественно по тримолекулярной реакции, что доказано Е.Т. Денисовым [7]. Перекисные радикалы в среде окисляющегося углеводорода могут не только взаимодействовать с компонентами реагирующей смеси (например, с исходным углеводородом), образуя гидроперекиси, но и подвергаться распаду с образованием стабильного продукта и нового свободного радикала, как это наблюдается при окислении углеводородов в газовой фазе[3]. Н.С.Ениколопяном показано, что в сложных цепных реакциях, протекающих с образованием ряда стабильных промежуточных продуктов, длина цепи может меняться по ходу реакции, что в свою очередь приведет к изменению скорости реакции, остановка окисления углеводородов задолго до полного расходования исходных веществ, постоянная скорость протекания реакции до очень больших глубин превращения (наблюдаемая для метана, бензола и др.) , несовпадение порядка реакции, определённого по ходу процесса, с определённым по начальной концентрации исходного углеводорода, автокатализ промежуточными и конечными продуктами, катализирующее и ингибирующее действие одних и тех же веществ в различных реакциях могут получить удовлетворительное объяснение в рамках представлений о том, что если в результате реакции стабильных промежуточных продуктов реакции с радикалом образуется радикал, более активный, чем исходный, то имеет место удлинение цепи. В противном случае по мере накопления стабильных промежуточных продуктов длина цепи уменьшается [39] . В условиях окисления гидроперекиси могут расходоваться не только при взаимодействии со свободными радикалами и по реакции разветвления, обычно протекающей медленно, но и другими путями, которые для общности называют не цепным расходованием. В некоторых реакциях окисления такой не цепной путь распада оказывается доминирующим. Так, при окислении альдегидов образующаяся над кислота реагирует с исходным альдегидом с образованием кислоты. В присутствии кислот гидроперекиси подвергаются гетеролитическому расщеплению, что приводит к автоторможению в реакции окисления. Работами Н.М.Эмануэля [3] показано, что ряд реакций окисления углеводородов прекращаются задолго до полного израсходования исходного вещества. Вопросы автоторможения реакций окисления подробно изучены Е.Т.Денисовым [40]. Было показано, что в начальный период окисление углеводорода осуществляется за счет взаимодействия RH с перекисными радикалами:
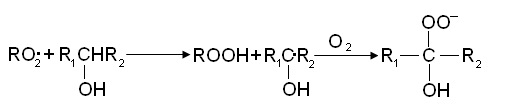
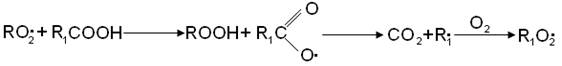
И скорость реакции определяется концентрацией перекисных радикалов. По мере накопления продуктов окисления - гидроперекисей, спиртов, кетонов, кислот - перекисные радикалы вступают в реакцию взаимодействия с этими продуктами. При цепном распаде вторичной гидроперекиси радикал R02 заменяется на свободный гидроксил: Реакция RO2• со спиртом приводит к образованию оксигидроперекисного радикала: Реакция с кислотами приводит к выделению СО2 и другому R1O2• радикалу:
Реакция RO2• со спиртом приводит к образованию оксигидроперекисного радикала:
Реакции с кислотами приводит к выделению СО2 и другому R1O2• радикалу:
В развившейся реакции исходный углеводород может взаимодействовать с различными свободными радикалами, и скорость его окисления зависит не только от общей концентрации радикалов, но и от их состава. В реальных процессах окисления скорость реакции по ходу изменяется не только в зависимости от скорости вырожденного разветвления цепей , но и в зависимости от состава радикалов. Специальными исследованиями и кинетическими расчетами было установлена [38], что в сложных цепных реакциях, протекающих с образованием молекулярных промежуточных продуктов, состав радикалов неизбежно меняется в ходе реакции вследствие изменения состава продуктов. Изменяющийся состав радикалов воздействует не только на суммарную активность радикалов, но и на их общую концентрацию путем изменения скорости квадратного обрыва цепей. Таким образом, механизм воздействия состава радикалов на скорость сложной цепной реакции таков, что ускорения реакции всегда ограничено, а её замедления может быть сколь угодно сильным. Это обстоятельство и является фундаментальной причиной широко распространенного явления самоторможения реакций окисления. Продукты распада радикала R02 были обнаружены при жидкофазном окислении н. бутана, изопропилбензола и циклогексана в металлических реакторах. Интересным представляется наблюдение [3]. О том, что в металлических реакторах продукты, образующейся с разрывом углеродного скелета в случае окисления бутана, составляют около 10-12% от прореагировавшего бутана, тогда как при окислении н. декане было обнаружено ни карбонильных соединений, ни спиртов, содержащих в своей молекуле меньшее число атомов углерода, чем исходный декан. Это на первый взгляд кажется удивительным, поскольку оба углеводорода принадлежат к одному и тому же классу- к парафинам нормального строения. В действительности никакого различия механизмом окисления н. декана и н. бутана не существует, а наблюдаемое расхождение в составе продуктов этих реакций объясняется, влиянием нержавеющей стали на распад радикала R-2 при проведении процесса в металлических реакторах. Под влиянием металла происходит также уменьшения периода индукции и увеличение суммарной скорости окисления, определяемой по скорости расходования бутана. При этом скорости накопления продуктов распада увеличиваются в несколько раз больше, чем скорости накопления продуктов гидроперекисного направления. Ускорение реакции связано, по-видимому, с увеличением скорости разветвления цепи за счет увеличения скорости распада гидроперекиси под действием металлической поверхности. Реакция распада радикала RO2• предшествует его изомеризация с переходом свободной валентности от атома кислорода к одному из соседних атомов в радикале. Изомеризация перекисного радикала происходит наиболее легко в случае, когда в третичные атомы углерода находятся в β - положении относительно друг - друга. При этом если свободная валентность, переходить к атому углерода, то вслед за изомеризацией радикала происходит разрыв связи С-С. Состав продуктов окисления сжиженного бутана при температуре 145°С и давлении 50 атм. В реакторе из нержавеющей стали отличается от состава продуктов окисления н. бутана в стеклянном реакторе. Наряду с соединениями, образующимися из гидроперекиси (метил этил кетон, вторичный бутиловый спирт, уксусная кислота), обнаруживается существенным количества веществ, содержащих меньшее число атомов углерода, чем исходный бутан (ацетальдегид, ацетон, метиловый и этиловый спирт и другие). Показано, что эти соединения не является продуктами дальнейшего превращения гидроперекиси, так как при термическом разложении гидроперекиси в атмосфере азота в тех же условиях, в которых проводится процесс окисления бутана, образуются только бутиловый спирт и метил этил кетон. Если изомеризация
радикала В реакциях окисления углеводородов гидроперекиси очень часто главные, но не единственные первичные молекулярные продукты окисления. Во многих случаях параллельно с гидроперекисями образуются циклические и полимерные перекиси, окиси и другие продукты окисления. Таким образом, из литературы известно, что металлы, контактирующие с окисляющимся углеводородом не всегда инертны к процессу окисления. Катализ процесса окисления солями металлов переменной валентности. При окислении углеводородов в качестве катализаторов обычно применяют органические соли кобальта, марганца, железа, меди, хрома, свинца, никеля. Перманганат калия, например, служит катализатором окисления парафина кислородом воздуха в производстве жирных кислот [2]. Катализаторы позволяют, проводит окисления при более низкой температуре, т.е. в более мягких условиях и таким образом уменьшают количество нежелательных продуктов глубокого окисления [3]. В реакциях окисления углеводородов механизм соленого катализа очень сложный. Ускоряя реакцию окисления, катализатор испытывает обратное воздействие продуктов окисления, что приводит к протеканию процесса в несколько последовательных стадиях. Каталитические действие соединений металлов переменной валентности указывает на цепной характер окисления. Изучение особенностей жидкофазного окисления углеводородов инициированного солями металлов, проведенное В.Г.Фрейдиным [80],показало, что период индукции при использовании двухвалентных металлов (Мn) значительно длиннее, чем при применении трехвалентных (Сr); период индукции увеличивается (в изученных пределах) с повышением содержания двухвалентного металла; спектры поглощения образующихся в индукционном периоде соединений металлов в высшем валентном состоянии соответствуют спектрами поглощения известных комплексных органических солей этих металлов; анализ стеарата кобальта, изменившегося и индукционном периоде окисления керосина; дает возможность приписать ему строение частично гидролизованного многоядерного комплексного соединения, присутствие спиртов ускоряет переход металлов в высшее валентное состояние [85]. Известно, что в зависимости от валентного состояния, ионы металлов переменной валентности могут присоединять или отдавать один электрон какой-либо валентно насыщенной молекуле. Это неизбежно приводит к образованию свободных радикалов, ускоряющих цепной процесс окисления.
Ион трех валентного металла в среде реакционной массы образует многоядерный катион:
Реакция с участием многоядерного катиона ускоряют реакцию и приводят к образованию продуктов окисления:
где: Ас- - анион кислоты (продукта окисления). Таким образом, в начале процесса окисления с участием двухвалентных ионов металлов переменной валентности замедление реакции объясняется обязательной последовательностью процессов: первичного инициирования, необходимая продолжительность которого увеличивается в результате большой потребности в первичных продуктов окисления (гидроперекисей), участвующих в образовании комплекса; реакции образования комплекса; процесса разрушения комплекса с образованием ионов и радикалов осколков комплекса, инициирующих развитую реакцию [31]. Было показано [4], что под действием кислорода эполеты металлов разлагаются, образуя две молекулы кислоты. Для практического использования катализатора большое значение имеет вопрос о стабильности жирных кислот в условиях технологического режима окисления. Тем не менее роль катализатора в процессе окисления высокомолекулярных жирных кислот выяснена недостаточно. Была изучена окисляемость фракций синтетических жирных кислот Сю -Ci3 и Си - С20. при переменном температурном режиме и в присутствии 0,2% КМпОд кислоты Сю - Сю окисляются незначительно, а кислоты Сю - Сго с большими скоростями. Кислотное число водорастворимых кислот по мере протекания каталитического окисления непрерывно повышается. Это свидетельствует о том, что кислоты обогащаются низкомолекулярными веществами. Наиболее эффективно процесс окисления ускоряется некоторой оптимальной концентрацией Мn, ровной -0,1%. Избыток КМnО4 по сравнению с оптимальной концентрацией или увеличение доли щелочного металла в составе катализатора приводят к разному уменьшению скорости процесса, в то время как один марганец влияет на скорость окисления гораздо слабее, чем в смеси с калием. Таким образом, основные ингибирующие функции в данном случае принадлежат, по-видимому, соединением щелочного металла [3]. Воздействие катализатора на реакцию окисления проявляется тем отчетливее, чем ниже температура окисления. При невысокой температуре катализированной окисления намного быстрее некатализированного. С повышением температуры различие в скоростях уменьшается. Это связано с тем, что предостаточно высокой температуре цепной процесс окисления способен к быстрому развитию в отсутствие катализатора, а солей катализатора выпадает в осадок на сравнительно неглубоких стадиях процесса вследствие накопления кислот и почти не участвует в реакции [4]. В промышленном производстве синтетических жирных кислот окисляют смесь (1:2) исходного парафина с возвратным, т.е. полученным после отделения продуктов реакции. Необходимая условия нормального протекания процесса присутствие катализатора. Обычно используют окиси марганца, содержащие щелочь или перманганат калия в количестве 0,08-0,1% от веса загрузки, считая на марганец. Реакция проходит при переменном температурном режиме 125-105°С. Постепенное снижение температуры по мере накопления продуктов окисления предотвращает обогащение жирных кислот побочными веществами, уменьшает концентрацию полифункциональных соединений окси кислот и т.п. Опытным путем было установлена, что окисление при более высокой постоянной температуре (125°С), хотя и значительно сокращает время реакции, но отрицательно сказывается на качестве синтетических жирных кислот. Процесс окисления прерывается при достижении кислотного число 70 [1]. Практическое осуществление окисления парафинов связано с использованием в этой реакции перманганата калия в качестве катализатора. Изучение непосредственного взаимодействия КМnO4 и MnO2 с парафином затрудняется тем, что эти катализаторы на начальных стадиях реакции находятся в гетерогенном состоянии. Экспериментальное изучения поведения KMnO4 и MnO2 в среде расплавленного парафина показало, что ни то, ни другое соединение без кислорода не взаимодействует с углеводородами [3]. Это свидетельствует о том, что реакция взаимодействий Мn+2 с радикалами RO2• конкурирует с реакцией продолжения цепи таким образом, что при уменьшении концентрации углеводорода до определенного значения наблюдается полное прекращение процесса окисления. Введение солей калия стабилизирует марганцовый катализатор и предотвращает выпадение осадка. Более того, добавление стеарата калия к осадку соединений марганца вызывает растворение последнего. Одновременно с этим изменяется и кинетика окисления, увеличивается скорость образования свободных кислот, снижается содержание карбонильных соединений в оксидате [32]. В результате анализа литературы видно, что процессы окисления углеводородов проводятся с участием солей металлов переменной валентности, которые улучшает условия образования кислот. Литературы 1.Каримов И.А., Мировой финансово – экономический кризис, пути и меры его преодолению в условиях Узбекистана / И.А.Каримов Т.: 2009.- 56 с. 2. Регламент производство синтетических жирных кислот / Волгаградский НПЗ. Волгоград, 1982 – 120с. 3. Эммануэль Н.И. Цепные реакции окисления углеводородов в жидкой фазе / Н.И. Эммануэль – М.: Наука, 1965 – 362 с. 4. Юкельсон И.И., Технология основного органического синтеза / И.И. Юкельсон. М.: Химия, 1968. – 672 с. 5.Эвери Г. Основы кинетики и механизма химической ркакций/ Г.Эвери; пер.с анг. В.В.Смирного – М.: Мир 1978 – 216 с. 6. Денисов Е.Т. Химическая кинетика / О.М. Саркисов, Г.И. Лихтенштейн . – М.: Химия 2000 – 568с. 7. Шипаева Т.А. Синтез и изучение свойств многофункциональных добавок на основе хлорпарафинов: дис.канд.хим.наук/ Шипаева Татьяна Александроовна. – Волгоград – 1998.-120 с. 8. Теоритическое изучение механизма окисления углеводородров молекуляном кислородом / Е.В. Николаева, А.Г. Шамов, Г.М. Хропковский и др.// Нефтехимия – 99; тез. докл V конф. по интенсификации нефтехимических процессов – Нижнекамск – 1999. – с. 103- 105. 9.Общая органическая химия : в 8 т. Т.1 / Д.Бартон, У.Д. Оллис; пер. с англ. С.В. Яроцкого; под ред. Н.К.Когеткова, А.И. Усова – М.: Химия 1982 – 856 с. 10. Oxidation of alkanes b TBHR in the presence of soluble titanum complexes / Fujewata Mashario, Xu Qiang, Souma Yoshie etc.// J.Hol.Catal. – 1999, - №1- P.77-84. 11. Meunier B.S.Oxidation catalysis: Pap. first international conference on porphyrins and phthalocyanines (ICPP-1)/ B.S. Meunier// Porphyrins and phthalocyanines -2000. -№4-P. 353 12. Пат. 6037507 США, МПК С 07 С 29/50. Oxidation process of branched aliphatic hydrocarbons and process for producing the oxide / Nakano Tatsuya, Isliii Yasutalca; заявитель и патентообладатель Daicel Chemical Ind. -№09/037703; заявл. 10.03.98; опубл. 14.03.00. ~3с. 13. Selective oxidation of n-butane on a V-P-O-catalyst: Improvement of the catalytic performance under fuel- rich condition by doping / S.Mota, J.C.Volla, G.Vorbeck. etc. //J.Chem. Soc- 2000.- 2.-P.319-329. 14. Catalytic Shilov chemistri: Plaiimnn chloride- catalyzed oxidation of terminal mctihyl groups by dioxigen /Lin Minren, Slien Chengyu, Garsia- Zayas Eduardo A. etc. // J. Amer.Chem, Soc.-2001. -№5. –P. 1000 -100L. 15. Laszio, J. Csanyl. Investigation of the catalytic behavior of ion-pair complexes of vanadium (5+) in the liquid- phase oxidation of hydrocarbons with molecular Oi / Csanyl Laszio J.Jaky Katoly, Galkaes Gabor // J.Mol. Catal. -2000, -№1-2. –P. 109-124. 16. Артемов, А.В. Новые высокоэффективные катализаторы жидкофазных окислительных процесс >> / А.В. Артемов // Катализ и промышленност. -2000.-№2.–С.18-23. 17. Заявка 19924533 Германия, МПК С 07С 57/07. Verfagen zur Marstcllung von Acrykaurc / Sclnfider Jiirdc, Ncstlcr Gerhard, Miiller- Enge J Klaus Joachim; заявитель и патентообладатель BASF AG.- №19924533.9; заявл. 28. 05. 99; Опубл. 30,11 02.-2с. 18. Заявка 19941315 Германия, МПК С 07 С 407/00. Selective oxidation von kohlenwasscretoffen / Langer Rein hard, Fengler Gerd; заявитель и иатентообладатель BASF AG.- №19941315.0, заявл. 31.08.99; опубл. 01.03.01. -3с. 19. Пат. 5981420 США, МПК В 01 J 31/00. Oxidation catalytic system and oxidation process / Nakano T. Isitt Y; заявитель и потентообладатель Daicel Chemical Ind. Ltd.; Yasutaka Isitt.-№09/024514; заявл. 17.02.98; опубл. 09.11.99.-с. 20. Заявка 19823088 Германия, МПК С 07 С 51/21. Verfahren zur HcistcUung Von Sauren / Riidinger Ch, Eberle H,-J., Bogner R., Kohlmarm W.; заявитель Consortium fur elektrocliemische Industrie GmbH.-№19823088.5; заявл. 22.05.98; опубл. 25.11.99.-4c. 21. Заявка 981066/04 Россия, МТЖ С07 В 41/08. Способ окисления углеводородов, спиртов и /или кетонов / Константины Мишель, Фаш Эрик, Родья Фибер Э.; заявитель Резон Эяермедиа, -№98106628/04; заявл. 09.04.98; опубл. 27.01.00.-6c. 22. Пат. 5914013 США, МПК С 07 В 33/00. Selective Thermal and fotooxidatkm of hydrocarbons in zeolites by oxygen / Frei Hein?., Blatter Fritz, Sun Hai; заявитель и патентообладатель. The Regents of the University of Colifomia.-№08/874,679; заявл. 13.06.97; опубл, 22.06.98.-fic. 23. Заявка 19746667 Германия, МПК С 07 С57/05. Verfahren den heterogen katalysierten Gasphascnoxidation von Propan zu Acrolein nnd/ odcr Acriylsaure / Jachow II , Tsnten A, Univerricht S, Arnold A; заявитель BASF AG.-№19746667.2, заявл. 23.01.99.-7c. 24. Kiyoshi, Otsuka. Селективное окисление с участием оксидов азота/ Otsuka Kiyoshi, Yamanaka Ichiro Shokubai // Catalysis and Catalysis,-1999,-№8. –P.606-612. 25. Farinas, E.T. Pliotoinduced oxidation of hydrocarbons with cobalt (111)- alkylperoxy completes / E.T. Farinas, C.V. Nguyen, F.K. Mascherak // Inorg. Chin. Actc. -1997.-Vol.263, 1-2,P. 17-21. 26. Светланов, КВ, Окисление алканов до карбоновых кислот / Н.В, Светланов, Е.А Николаева //Научная сессия : аннотац. сообщ./ 1СГТУ.-Казань, 2003.-с.34. 27. Пат, 6340420 США, МГЖ В 01 D 61/44. Methods of treating the oxidation mixture of hydrocarbons to respective dibasic acids / Dassel Mark W, Vassiliou Euslathics; заявитель и патентообладатель 09/3458S0; заявл. 30.06.99; опубл. 22.01 т.-Лс. 28. Шт. 6515146 США, МШС С 07 D 307/60, C 07 C 51/16. Process for catalytic selective oxidation of hydrocarbon substrate / Perrcgaard Jens Santamaria Jesus, Menendes Miguel etc; заявитель и патентообладатель Mai dor Topsoe A/S, University of Zaraoza, Du Pout Iberia S.A.-fe 09/654299; заявл. 01.09.00", опубл. 04.02.03.-бс. 29. Заявка 282S194 Франция, МПК С 07 С 51/13, Procede d'oxydation d'hydrocarbures en acides / Bonnet Didicr, Fache Brie, Simotiato Jean Pierre; заявитель P.Jiodia Polyamide Intermediates SAS.-N 0110427; заявл. 03.08.01; опубл. 07.02.03.-4с. 30. Shulpin, G.B. Melall- catalysed hydrocarbon oxygenations in solution: The dramatic role of additives: A review / G.B. Shulpin // J.Mol Calal.A, -2002,-№1.-P.39-66. 31. Заявка 10201241 Германия, МПК В 01 J 31/02. Katalysator / Weisbeck Markus, Mcincii Maric- Therese, Schmirt Jurg etc.; заявитель Bayer AG. -№ 10201241.5; заявл. 15.01.02; опубл. 24.07.D3.-6c. 32. Заявка 2824322 Франция, МГЖ С 07 С 037/00. Precede d'oxydation d'hydrocarbures / Fache Eric, Simonato Jean Prierre (RHODIA SERVICES ); зритель RHODIA POLYAMIDE INTERMEDIATES SAS.- 0106016; заявл. 04.05.01; опубл. 08.11.02.-6с. 33. Заявка 200110090/04 Россия, МПК С 07 С 51/43. Способ выделения и очистки карбоновой кислоты, образующиеся при реакции прямого окисления углеводородов / Константин и Мишель, Фаш Эрик, Маремм Шильбстр; заявитель Родиа Полиамид Интермедиа. – № 200110090/04; заявл.14.04. 99, опубл. 20.01.03.- 4 с. 34. Заявка 19622331 Германия, МПК С 07 С 47/22. Verfahren der Heterogen katalisierten Gaspasenoxidation von Propan zu Acrolein / Tenten A., Proll Th., Schildberg M.-P.; заявитель BASF AG. -№ 19622331.8; заявл. 04.06.96; опубл. 11.12.97.- 4 с. 35. Пат. 5536875 США, МПК С 07 С 51/16. Enhanced oxidation of organic chemicals / Roby Anne K., Kingsley Jeffrey P.; заявитель и пантенто- обладатель Praxair Technology Inc.; заявл. 22.05.95; опубл. 16.07.96.- 3с. 36. Заявка 2732678 Франция, МКИ С 07 С 55/14, С 07 С 51/215. Procede d’oxydation d’hydrocarbures, d’alcogols ou de cetones par catalyse heterogene / Custantini M., Fashe E., Gilbert L.; заявитель Rhone-Paulene Chimie. - №9504428; заявл. 07.04.95; опубл. 11.10.96. – 3 с. |
|
|
| 17.06.2012 |
| Большое обновление Большой Научной Библиотеки |
| 12.06.2012 |
| Конкурс в самом разгаре не пропустите Новости |
| 08.06.2012 |
| Мы проводим опрос, а также небольшой конкурс |
| 05.06.2012 |
| Сена дизайна и структуры сайта научной библиотеки |
| 04.06.2012 |
| Переезд на новый хостинг |
| 30.05.2012 |
| Работа над улучшением структуры сайта научной библиотеки |
| 27.05.2012 |
| Работа над новым дизайном сайта библиотеки |